Software
The Nordenfelt Lab publishes its code on Github
Code related to publications:
Binding site localization on non-homogeneous cell surfaces using topological image averaging
A method to carefully measure the molecular distance between a site of interest and a reference surface by the repeated acquisition of the two image channels followed by the statistical calculation of the relative difference.
Publication: eLife 2022;11:e64709 | doi: 10.7554/eLife.64709
Download:

A predictive model of antibody binding in the presence of IgG-interacting bacterial surface proteins
Many bacteria can interfere with how antibodies bind to their surfaces. This bacterial antibody targeting makes it challenging to predict the immunological function of bacteria-associated antibodies. The M and M-like proteins of group A streptococci exhibit IgGFc-binding regions, which they use to reverse IgG binding orientation depending on the host environment. Unraveling the mechanism behind these binding characteristics may identify conditions under which bound IgG can drive an efficient immune response. Here, we have developed a biophysical model for describing these complex protein-antibody interactions. We show how the model can be used as a tool for studying various IgG samples’ behaviour by performing in silico simulations and correlating this data with experimental measurements. Besides its use for mechanistic understanding, this model could potentially be used as a tool to predict the effect, as well as aid in the development of antibody treatments. We illustrate this by simulating how IgG binding in serum is altered as specified amounts of monoclonal or pooled IgG is added. Phagocytosis experiments link this altered antibody binding to a physiological function and demonstrate that it is possible to predict the effect of an IgG treatment with our model. Our study gives a mechanistic understanding of bacterial antibody targeting and provides a tool for predicting the effect of antibody treatments.
Publication: Front. Immunol., 22 March 2021 | doi: 10.3389/fimmu.2021.629103

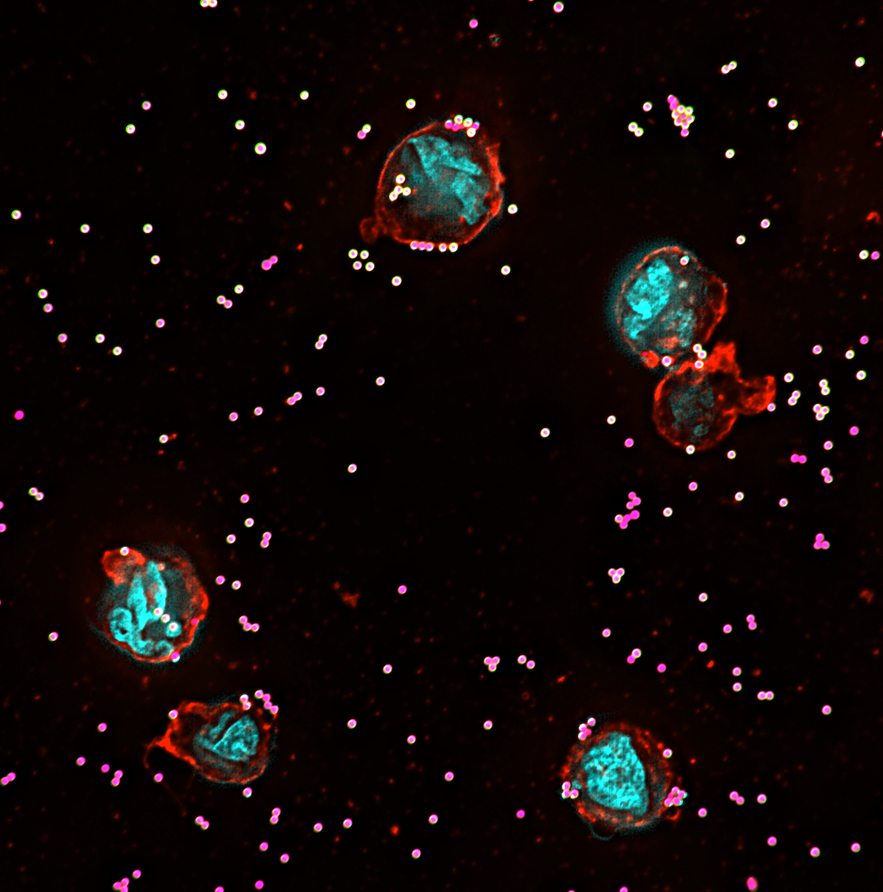
High-sensitivity assessment of phagocytosis by persistent association-based normalization
Phagocytosis is measured as a functional outcome in many research fields but can be challenging to quantify accurately, with no robust method available for cross-laboratory reproducibility. Here, we identified a simple, measurable parameter – persistent prey-phagocyte association – to use for normalization and dose-response analysis. We apply this in a straightforward analytical method, persistent association-based normalization (PAN), where first the multiplicity of prey (MOP) ratio needed to elicit half of the phagocytes to associate persistently (MOP50) is determined. MOP50 is then applied to normalize for experimental factors, with adhesion and internalization analyzed separately. We use THP-1 cells and different prey and opsonization conditions to compare the PAN method to standard ways of assessing phagocytosis, and find it better in all cases, with increased robustness, sensitivity, and reproducibility. The approach is easily incorporated into most existing phagocytosis assays and allows for reproducibly comparing results across experiments and laboratories with high sensitivity.
Link to paper in The Journal of Immunology
Analysis template download links to GitHub repository
NETQUANT – automated quantification of neutrophil extracellular traps
Neutrophils can eject extracellular traps (NETs) that are extensive webs of DNA covered with antimicrobial proteins into the extracellular environment during infection or inflammation as a part of their defense arsenal. Image acquisition of fluorescently labeled NETs and subsequent image-based quantification is frequently used to analyze NET formation (NETosis) in response to various stimuli. However, there are important limitations in the present methods for quantification. Manual methods tend to be error-prone, tedious and often quite subjective, whereas the software-rooted options are either semi-automatic or difficult to operate. Here we present an automated and uncomplicated approach for quantifying NETs from fluorescence images, built as a freely available app for MATLAB®. It is based on detection of a set of clearly defined parameters, all related to the biological manifestation of NETs and allowing for single-cell resolution quantification and analysis.
Link to paper in Frontiers in Immunology
Download:

![]()
Matrix-masking to balance nonuniform illumination in microscopy
With a perfectly uniform illumination, the amount and concentration of fluorophores in any (biological) sample can be read directly from fluorescence micrographs. However, non-uniform illumination in optical micrographs is a common, yet avoidable artefact, often caused by the setup of the microscope, or by inherent properties caused by the nature of the sample. In this paper, we demonstrate simple matrix-based methods using the common computing environments MATLAB and Python to correct nonuniform illumination, using either a background image or extracting illumination information directly from the sample image, together with subsequent image processing. We compare the processes, algorithms, and results obtained from both MATLAB (commercially available) and Python (freeware). Additionally, we validate our method by evaluating commonly used alternative approaches, demonstrating that the best nonuniform illumination correction can be achieved when a separate background image is available.
Link to paper in Optics Express
Matrix-masking download links: